5.4. Aerosols#
Formation, growth, composition, and removal processes
Key Points:
The atmosphere contains numerous aerosol particles covering a size range of \(1-2 \ \mathrm{nm}\) up to \(\gt 100 \ \mathrm{\mu m}\) diameter and containing a variety of chemical constituents. Particle distribution is divided into a nucleation, an Aitken, an accumulation, and a coarse mode.
Primary particles are directly emitted into the atmosphere. Secondary particles are formed in the atmosphere from gaseous precursors through nucleation.
Primary sources of particles and their gaseous precursors can be natural or anthropogenic.
Particles are removed from the atmosphere through dry and wet deposition.
Aerosol chemical composition varies widely depending on the sources and location. Main components are nitrate, sulphate, chloride, ammonium, sodium, potassium, calcium, and organic material.
Heterogeneous reactions occur on the surface or in the condensed phase of aerosol particles. This leads to the chemical conversion of trace gases and their removal. The rate of this process is usually expressed in terms of the uptake coefficient, \(\gamma\), defined in terms of kinetic theory of gases.
Introduction to atmospheric particles#
The atmosphere also contains a wide variety of microscopic particulate material, known as the atmospheric aerosol or particulate matter (PM). The aerosol plays an important role in both the physics and chemistry of the atmosphere.
Atmospheric aerosol particles may be liquid, solid or multiphase. In the stratosphere the aerosol consists predominantly of sulphuric acid droplets formed by oxidation of sulphur gases (\(\mathrm{SO_2}\) and \(\mathrm{OCS}\) (carbonyl sulfide)). In the troposphere the aerosol is much more complex.
Aerosol particles are absorbing and/or scattering short wave radiation and are therefore directly influencing the radiative balance of the atmosphere. The intensity of light scattering or absorption is strongly dependent on the size and chemical composition of particles.
Aerosols are also involved in the formation of clouds - the water droplets that comprise clouds always condense on certain pre-existing aerosols called cloud condensation nuclei. Particle size and chemical composition are key parameters, which determine whether an aerosol particle is an efficient nucleus for cloud droplet formation. These aerosol particle-cloud interactions lead to a complex feedback system influencing the hydrological cycle.
Both processes, light absorption/scattering and could formation, are not very well understood on a global level and are one on the main uncertainties associated with studies predicting the future climate. The uncertainty is mainly due to the large variability in the physical and chemical properties of aerosol particles, as well as their temporal and spatial distributions.
Aerosols also provide a sink for traces gases. The reactions of gaseous compounds at the surface or within the condensed phase of aerosol particles are called heterogeneous reactions.
Particles are involved in health impacts caused by air pollution. Aerosol particles \(\lt 10 \ \mathrm{\mu m}\), which penetrate into the lung (larger particles are deposited in the nose and mouth), have been shown to correlate with increasing rates of respiratory and cardio-vascular diseases in epidemiological studies. The particle properties causing these health effects are not well understood, but particle size and the chemical composition (e.g., transition metals, oxidizing organic components) are likely responsible for the observed particle toxicity.
Size distribution of atmospheric aerosols#
The size of atmospheric particles ranges from \(\lt 1-2 \ \mathrm{nm}\) to \(\gt 100 \ \mathrm{\mu m}\) diameter. A typical distribution as a function of size is shown in Fig. 5.15. The distribution is usually divided into the nucleation mode (\(\lt 10 \ \mathrm{nm}\)), the Aitken mode (\(\gt 100 \ \mathrm{nm}\)), the accumulation mode (from \(100 \ \mathrm{nm}\) to \(1-2 \ \mathrm{\mu m}\), and the coarse mode (\(\gt 1-2 \ \mathrm{\mu m}\)).
The maximum in the particle concentration generally lies in the nucleation mode size range when particles are measured in number per unit volume of air. The nucleation mode particles, although numerous, contribute very little to the aerosol mass.
The maximum in surface area, which is the important parameter for light scattering and for many heterogeneous reaction rates, usually lies in the so-called ‘accumulation’ mode. The maximum in volume/mass of particles per unit volume of air lies in the accumulation/coarse fraction.
The total particle number concentration varies greatly in the troposphere with \(\lt 20 \ \mathrm{cm}^{-3}\) for the polar regions and \(\gt 10^5 \ \mathrm{cm}^{-3}\) for an urban aerosol. The total number concentration decreases strongly with altitude and at altitudes of \(3-5 \ \mathrm{km}\) typical concentration of about \(100 \ \mathrm{particles} \, \mathrm{cm^{-3}}\) are found.
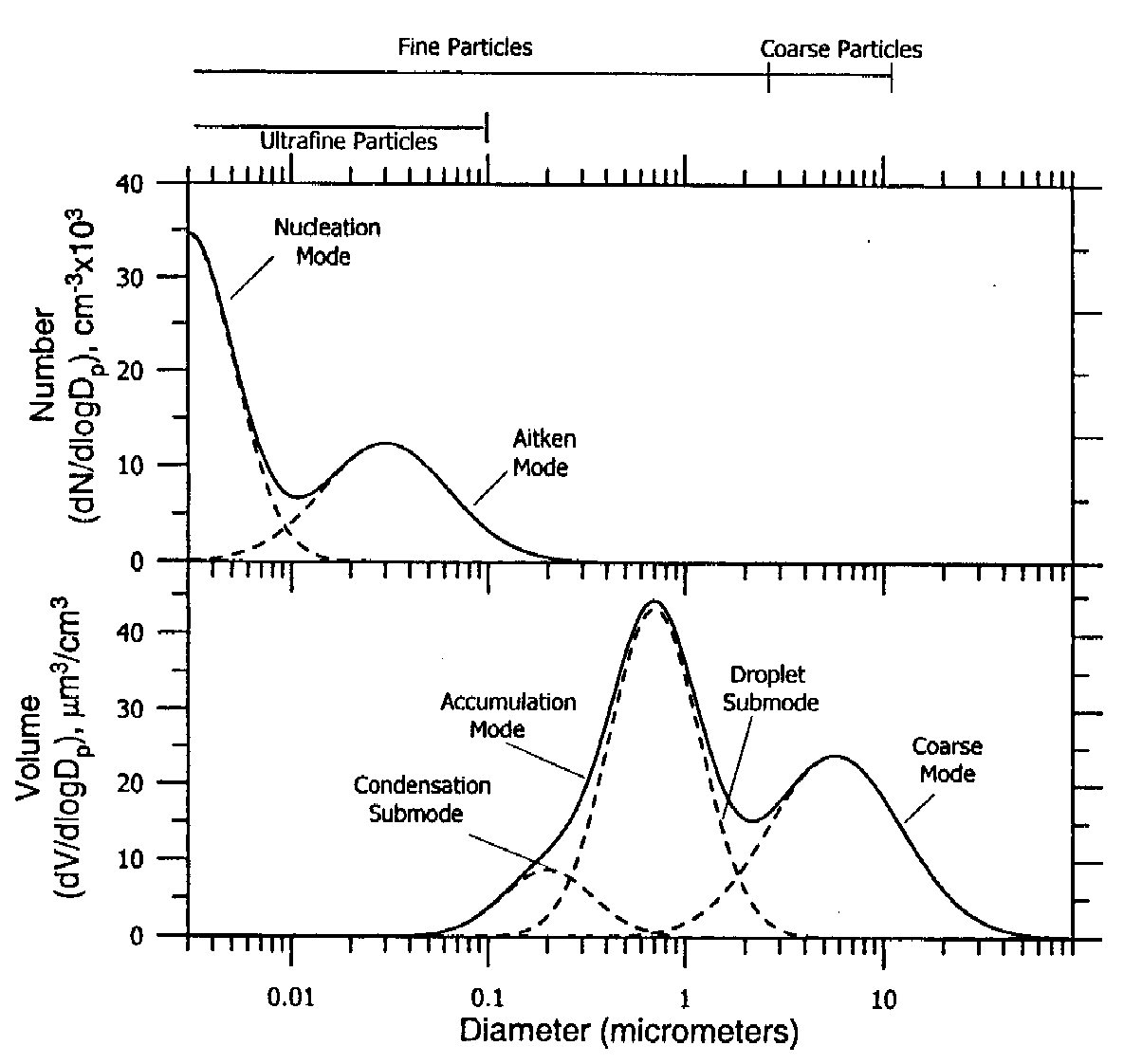
Fig. 5.15 Typical size distribution of the tropospheric aerosol.#
Formation processes and sources#
Formation processes and processes affecting size distribution#
Formation processes of aerosols are highly variable in time and space, which is one of the main reasons why the influence and interactions of particles with other parts of the climate system are so difficult to account for in atmospheric chemistry and climate models.
Particles in the coarse mode are usually produced mechanically by weathering and wind erosion processes or through bubble bursting over the ocean. Direct anthropogenic emissions contribute to coarse mode particles through traffic and industrial processes.
In the smallest size fraction (nucleation mode), particle formation arises via homogeneous nucleation, which is the formation of new particles through gas-to-particle conversion processes. Particle nucleation is frequently observed over land mainly in remote areas but also over the ocean. Chemical compounds forming the initial clusters of atmospheric nucleation particles are not well characterized but water and sulphuric acid, ammonium, amines, and highly oxygenated organic molecules (HOMs) are most likely important in forming the initial nucleation clusters. Organic compounds are thought to condense onto the initial \(\mathrm{H_2O}\) - \(\mathrm{H_2SO_4}\) clusters, i.e., being responsible for the initial growth of the clusters into stable particles of a few \(\mathrm{nm}\) in size (see Fig. 5.16).
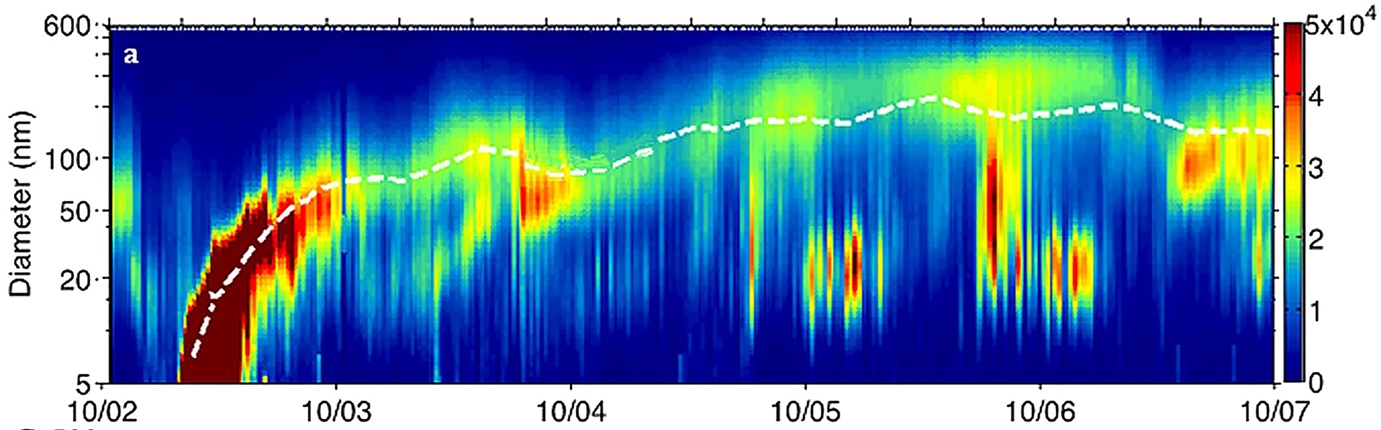
Fig. 5.16 Typical evolution of particle size distribution after a particle nucleation event (Beijing 2—7 October 2013). White dashed curve represents the mean diameter. From Guo et al 2014, Lee et al 2019.#
The high initial number concentration of particles with \(d < 10 \ \mathrm{nm}\) are rapidly reduced through coagulation, resulting in aerosol lifetimes of the order of minutes for these smallest particles. The nucleation mode particles, although numerous, contribute very little to the aerosol surface or mass.
The coagulation rate of particles with size 1 and 2 is given by
where \(K_{1,2}\) is the coagulation coefficient, \(D\) is the diffusion constant, and \(d\) is the particle diameter.
\(d_1 \ (\mathrm{\mu m})\) \ \(d_2 \ (\mathrm{\mu m})\) |
0.01 |
0.1 |
1.0 |
10.0 |
|---|---|---|---|---|
0.01 |
67 |
|||
0.1 |
180 |
8.6 |
||
1.0 |
1700 |
24 |
3.5 |
|
10.0 |
16000 |
220 |
10.3 |
3.0 |
Particles in the accumulation mode typically arise from either: i) the condensation of low volatility vapours onto existing smaller particles, or ii) from coagulation of particles. Chemical modification (heterogeneous reactions) following condensation of semi-volatile components from the gas phase onto existing particles is another process increasing the mass of accumulation mode particles. The main removal process is incorporation into cloud droplets and subsequent wet deposition.
Particle sources#
Atmospheric aerosols may originate from either naturally occurring or anthropogenic processes. Further classification within each category into primary and secondary sources may also be made. Aerosols directly emitted into the atmosphere constitute primary sources while secondary sources arise from the gas-to-particle conversion of gaseous precursor compounds such as \(\mathrm{NO}_x\), \(\mathrm{SO_2}\) and volatile organic compounds (VOCs).
Natural sources#
Primary sources#
Sea Salt Sea salt aerosol results from the bursting of bubbles, formed by wave and wind-action at the ocean surface. Sea salt aerosol mainly consists of \(\mathrm{NaCl}\), \(\mathrm{KCl}\), \(\mathrm{CaSO_4}\), and \(\mathrm{Na_2SO_4}\). Soluble and insoluble organic compounds may also be important components.
Mineral Dust Wind-blown mineral dust (generated in physical and chemical weathering processes of rock and soil) from desert and semi-arid regions is an important source of tropospheric aerosols. Atmospheric size distributions of mineral dust particles range up to several hundred \(\mathrm{\mu m}\). The principal elemental constituents of mineral dust are oxides and carbonates of \(\mathrm{Si}\), \(\mathrm{Al}\), \(\mathrm{Ca}\), and \(\mathrm{Fe}\).
Primary Organic Aerosols/Biological Debris. Continental sources mainly arise from vegetation (plant waxes and fragments, pollen, spores, fungi, and decaying material), while marine sources consist of organic surfactants formed via bubble bursting. These particles are predominantly in the coarse mode.
Volcanic Emissions. Volcanic activity and particle emissions occurs on a sporadic basis. These emissions are composed of ash (principally \(\mathrm{SiO_2}\), \(\mathrm{Al_2O_3}\), and \(\mathrm{Fe_2O_3}\)), gases (\(\mathrm{SO_2}\), \(\mathrm{H2_S}\), \(\mathrm{CO_2}\), \(\mathrm{HCl}\), \(\mathrm{HF}\)), and water vapour. \(\mathrm{SO_2}\) emissions may contribute significantly to particle emissions of volcanic eruptions through oxidation reactions in the atmosphere leading ultimately to sulphuric acid.
Secondary sources#
Secondary natural aerosols may be formed from a number of natural precursor gas sources, containing sulphur, nitrogen and hydrocarbons. A main natural source is the release of gaseous dimethyl sulphide (DMS) from the oceans. DMS is formed from the biological activity of phytoplankton and eventually forms aerosol sulphate via the photo-oxidation to methanesulfonic acid and \(\mathrm{SO_2}\).
The formation of nitrate aerosols from nitrogen precursor gases has two main natural sources: i) \(\mathrm{NO_X}\) from lightning and soils, and ii) \(\mathrm{N_2O}\) from bacterial activity in soils and the oceans. Secondary organic aerosols (SOA) are formed by oxidation products of VOCs.
Anthropogenic sources#
Primary sources#
Biomass Burning. Natural wild fires and anthropogenic fires (e.g., for agricultural clearing) are commonly termed as biomass burning. Release of biomass burning products into the atmosphere occurs mainly in the tropics during the dry seasons.
Industrial Processes. Aerosol emissions from industrial processes have a diverse number of sources.
Soot. Soot is a ubiquitous component of the atmospheric aerosol. As a result of it being chemically relatively inert and having poor hygroscopic properties, it may be used as an anthropogenic tracer. Soot aerosols are generated in incomplete combustion processes and consist mainly of an organic fraction and an inorganic graphite-like carbon fraction (also called black carbon or elemental carbon).
Secondary sources#
Sulphate and Nitrate. \(\mathrm{SO_4^{2—}}\) and \(\mathrm{NO_3^—}\) are major components of tropospheric aerosol. They are formed from gaseous precursors (\(\mathrm{SO_2, \ NO_2}\)) through oxidation reactions mainly with OH in the gas phase or condensed phase leading to \(\mathrm{H_2SO_4}\) and \(\mathrm{HNO_3}\), respectively. Oxidation of \(\mathrm{SO_2}\) always results in the formation of aerosol mass, due to the very low \(\mathrm{H_2SO_4}\) vapour pressure, and is in contrast to \(\mathrm{HNO_3}\) which is distributed between the gas and aerosol phases. The largest source of \(\mathrm{SO_2}\) and \(\mathrm{NO_x}\) are fossil fuel combustion processes. Over continental surfaces where gaseous ammonia is present, \(\mathrm{H_2SO_4}\) forms \(\mathrm{NH_4HSO_4}\) and \(\mathrm{(NH_4)_2SO_4}\). In the presence of \(\mathrm{NH_3}\), \(\mathrm{HNO_3}\) (one of the major \(\mathrm{NO_x}\) reservoir compounds) forms \(\mathrm{NH_4NO_3}\).
\[\begin{split}\\ \mathrm{NH_4NO_{3~(s)} \longrightarrow NH_{3~(g)} + HNO_{3~(g)}}\end{split}\]Ammonia plays an important role in the neutralisation of acid species (\(\mathrm{HNO_3,\ H_2SO_4}\)), as it is the most common atmospheric alkaline gas. Major natural and anthropogenic sources respectively include:
soils and organic decomposition
fertilisers and animal farming
catalysed vehicles.
Secondary Organic Aerosol. Fossil fuel consumption and biomass burning, caused by human activities, are the main sources of anthropogenic VOCs that can lead to SOA. On a global scale, biogenic emissions from terrestrial plants and marine algae are dominant. SOA are formed by oxidation products of VOCs (in this context called ‘aerosol precursors’) in the atmosphere and thus contrast in their formation process compared to primary particles. These oxidation reactions are mainly initiated by reactions with ozone, \(\mathrm{OH}\), and \(\mathrm{NO_3}\) radicals. The ability of a VOC to form SOA depends on the volatility of its oxidation products, the concentration, and the reactivity of the VOC in the atmosphere.
Removal Processes#
Dry Deposition#
Removal at the Earth’s oceanic and terrestrial surfaces (water, soil and vegetation) in the absence of precipitation is termed ‘dry deposition’. The rate of this process is controlled by transport through the atmospheric boundary layer and by reaction or adsorption at the surface (i.e., depending on chemical/physical properties of the surface and the deposited particle or gas). If surface removal is efficient a gradient in concentration in the surface layers results.
The flux of a trace gas to the surface, \(F\), and its concentration \(c\), as measured at the same height above the surface, are related by the deposition velocity, \(v_g\):
The lifetime of a trace gas with respect to deposition is related to the deposition velocity and the height, \(h\), to which the trace gas is mixed by the equation:
For trace gases which are removed efficiently at the surface, e g. \(\mathrm{SO_2,~HNO_3,~O_3}\) (land surfaces only), values of \(v_g\) are typically of the order of \(\mathrm{1~ cm~ s^{-1}}\). \(\mathrm{SO_2}\) emitted from surface sources is mainly contained in a well-mixed boundary layer of height \(\mathrm{\sim 1~km}\). Its mean deposition velocity for North West Europe is \(\mathrm{\sim 0.8~cm~ s^{-1}}\), therefore the mean lifetime with respect to dry deposition is approximately 1.4 days.
Ozone has a similar deposition velocity over land, but has an approximately uniform concentration and can be considered well mixed up to the tropopause at \(\mathrm{12 \ km}\) altitude. The lifetime of tropospheric ozone with respect to deposition is therefore much longer, approximately 17 days.
Aerosol particles are also removed by dry deposition. Their deposition velocity is strongly dependent of the particle size. Particles below about \(\mathrm{50 \ nm}\) diameter behave like gases with respect to dry deposition and are efficiently removed due to Brownian diffusion. Particles in the range of \(\mathrm{2 - 20 \ \mu m}\) are mostly deposited due to inertial impaction and very large particles \(\mathrm{> 20~\mu m}\) are removed due to gravitational settling (sedimentation). As a result, the deposition velocity for airborne particles shows a minimum of \(\mathrm{0.01~cm~s^{-1}}\) for the particles of \(\mathrm{\sim 500~nm}\) (\(\mathrm{0.5~\mu m}\) – Fig. 5.17). Thus, aerosols in the range \(\mathrm{100 – 2000~nm}\) have the longest atmospheric lifetime and are termed the ‘accumulation mode’.
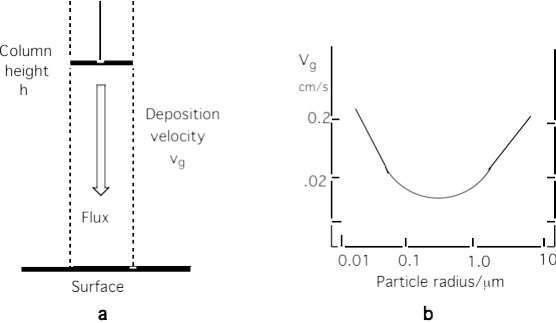
Fig. 5.17 a) Schematic illustration of surface deposition; b) size dependant deposition velocity of aerosol particles.#
Wet Deposition#
Wet deposition describes removal in the precipitation elements, cloud, rain, and snow. The process involves the incorporation of gases and particles into cloud droplets that then grow and eventually rain or snow out. This ‘rain out’ process is also known as ‘in-cloud scavenging’. Soluble gases enter the droplets by absorption that may be followed by chemical reaction. Aerosol particles may be incorporated into cloud drops by acting as condensation nuclei, around which the aqueous or ice particles grow and by coagulation with existing droplets.
The solubility is described in terms of the Henry’s law constant \(H_X~\mathrm{(M~atm^{—1}})\) which for species \(X\) the vapour pressure in the gas phase \(P_X\) is related to the liquid phase concentration \(\mathrm{[X]}_{liquid}\) by:
Values of \(H_X\) cover a large range (\( < 0.1 - 105 \mathrm{M \ atm^{—1}}\)).
A second mechanism involves the uptake into falling precipitation, termed ‘wash out’, or ‘below-cloud scavenging’. Removal times by wash out are of the order of a few hours for water-soluble gases in moderate rain. Because of the highly variable nature of precipitation events, quantitative estimation of overall wet deposition rates is difficult. As an approximation, the rate of wet deposition is taken as \(\mathrm{\lambda [X]}\) where \(\mathrm{[X]}\) is the concentration of the gas or aerosol and \(\lambda\) is the washout (scavenging) coefficient, which is proportional to the seasonal or annual mean precipitation amount and is typically of the order of \(0.12 - 0.5 \ \mathrm{day^{-1}}\) (a lifetime of 2 to 8 days).
Most processes involved in wet deposition are reversible, e.g., aerosol particles can be incorporated into a cloud droplet, which may evaporate again producing new particles. Equally, trace gases absorbed into particles can evaporate from the particle due to changes of the particle bulk composition (e.g., pH) or due to chemical reactions.
Aerosol Chemical Composition#
The chemical particle composition can be divided into 5 main categories: inorganic salts, metals, organic compounds, elemental (graphite-like) carbon and water.
Most abundant inorganic salts are \(\mathrm{(NH_4)_2SO_4}\) and \(\mathrm{(NH_4)NO_3}\) and over the ocean or in areas close to oceans also \(\mathrm{NaCl}\). \(\mathrm{(NH_4)_2SO_4}\) and \(\mathrm{(NH_4)NO_3}\) are formed via secondary processes in the atmosphere, whereas \(\mathrm{NaCl}\) is a primary particle component.
A large number of metals (exclusively from primary sources) is found in aerosol particles; however, they are often only a small fraction (<10%) of the total submicron particle mass. At larger diameters (i.e., the coarse mode) and especially in arid areas metals may become much more dominant. The most abundant metals found in aerosols are \(\mathrm{Al}\), \(\mathrm{Si}\) (metalloid), \(\mathrm{Mg}\), \(\mathrm{K}\), \(\mathrm{Ca}\), \(\mathrm{Ti}\), \(\mathrm{V}\), \(\mathrm{Fe}\), and \(\mathrm{Zn}\). Metals have been suggested to be important in the negative health effects of particle.
Heterogeneous reactions in the atmosphere#
In order to explain chemical change in the atmosphere we have to take into account, heterogeneous reactions occurring in/on the condensed phase.
Cloud droplets and aerosol particles provide an important medium for chemical reactions in the atmosphere.
Water soluble substances, e.g., \(\mathrm{SO_2}\), acids, \(\mathrm{H_2O_2}\), transfer readily to aqueous airborne particles, where they are adsorbed, solvated, and ionised. Hydrolysis in the condensed phase is an important reaction leading e.g., to formation of nitric acid from \(\mathrm{N_2O_5}\), which is a key process for the \(\mathrm{NO_x}\) budget. Acid-base reactions can lead to formation of non-volatile species.
Reduced substances can be oxidised by liquid phase oxidants, an important example being \(\mathrm{SO_2}\) which is converted to sulphuric acid in cloud droplets. Redox reactions involving hydrogen halides and halogen nitrates, which occur on ice particles are important in stratospheric chemistry, because they convert reservoir species to reactive forms of halogen which lead to ozone loss. Redox reactions may also be important for release of halogens from sea spray aerosols.
Some examples of important heterogeneous reactions:
Rates of heterogeneous reactions and resistance model of gas uptake#
Heterogeneous chemistry in the atmosphere involves a series of processes, including diffusion, accommodation (sticking), and evaporation from the surface. In the case of liquids, diffusion and dissolution will occur in the condensed phase. Chemical reaction may also happen at the gas-particle interface or in the condensed phase. These must be treated together to understand the overall rate of removal from the gas phase, as any one process can act in a rate determining way. The processes for liquid particles are represented in Fig. 5.18.
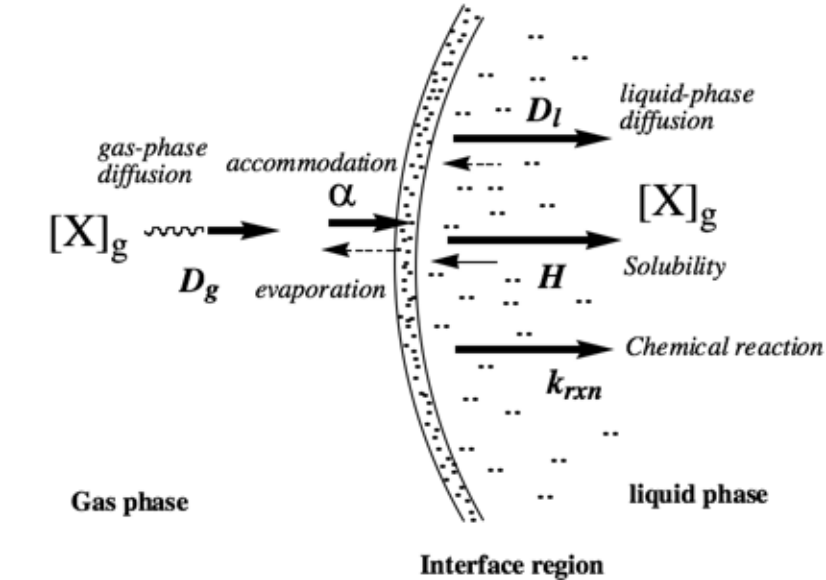
Fig. 5.18 Processes involved in transfer of molecules from the gas to the liquid phase#
The rate of a heterogeneous process is usually expressed in terms of the uptake coefficient, \(\gamma\), which is defined as the fraction of molecules colliding with the surface that are permanently lost from the gas phase.
According to the kinetic theory of gases, the rate of collision of a species \(\mathrm{A}\) at the surface, per unit area, is \(\frac{\mathrm{[A]} c}{4}\), where \(\mathrm{[A]}\) is the concentration (\(\mathrm{molecules~cm^{-3}}\)) of species \(\mathrm{A}\), and \(c\) the mean molecular speed of the gas molecules (\(c = (8 R T/ \pi M)^{1/2}\)).
The rate of removal of molecules at the surface can be expressed as a first-order process with rate:
where the first-order rate constant, \(k_{het}\), is defined as:
where \(S\) is the surface area of the atmospheric particles (N.B. per unit volume of gas, often called the surface area density and has units of e.g., \(\mathrm{cm^2~cm^{-3}}\)). As well as providing data on the rate of removal of trace gases by aerosol particles, uptake coefficient measurements can be used to deduce the mechanisms of the heterogeneous processes.
A useful approach for dealing with general problems of uptake rate into clouds and aerosols is the resistance model in which each process (gas transport, surface accommodation, solubility, chemical reaction) is expressed as a resistance term. Resistances are expressed as the inverse of the dimensionless uptake coefficients. The resistances \(1/\gamma_g\) and \(1/\alpha\) represent the resistance due to gas transport and surface accommodation respectively, and \(\gamma_{sol}\) and \(\gamma_{rxn}\) are associated with liquid solubility and chemical reaction respectively. On the basis of the model, the overall uptake coefficient is expressed as:
where \(\gamma\) is the experimentally determined overall net gas uptake coefficient.